
Help
Browse the vignettes (use arrows or swipe). Click an image to zoom.
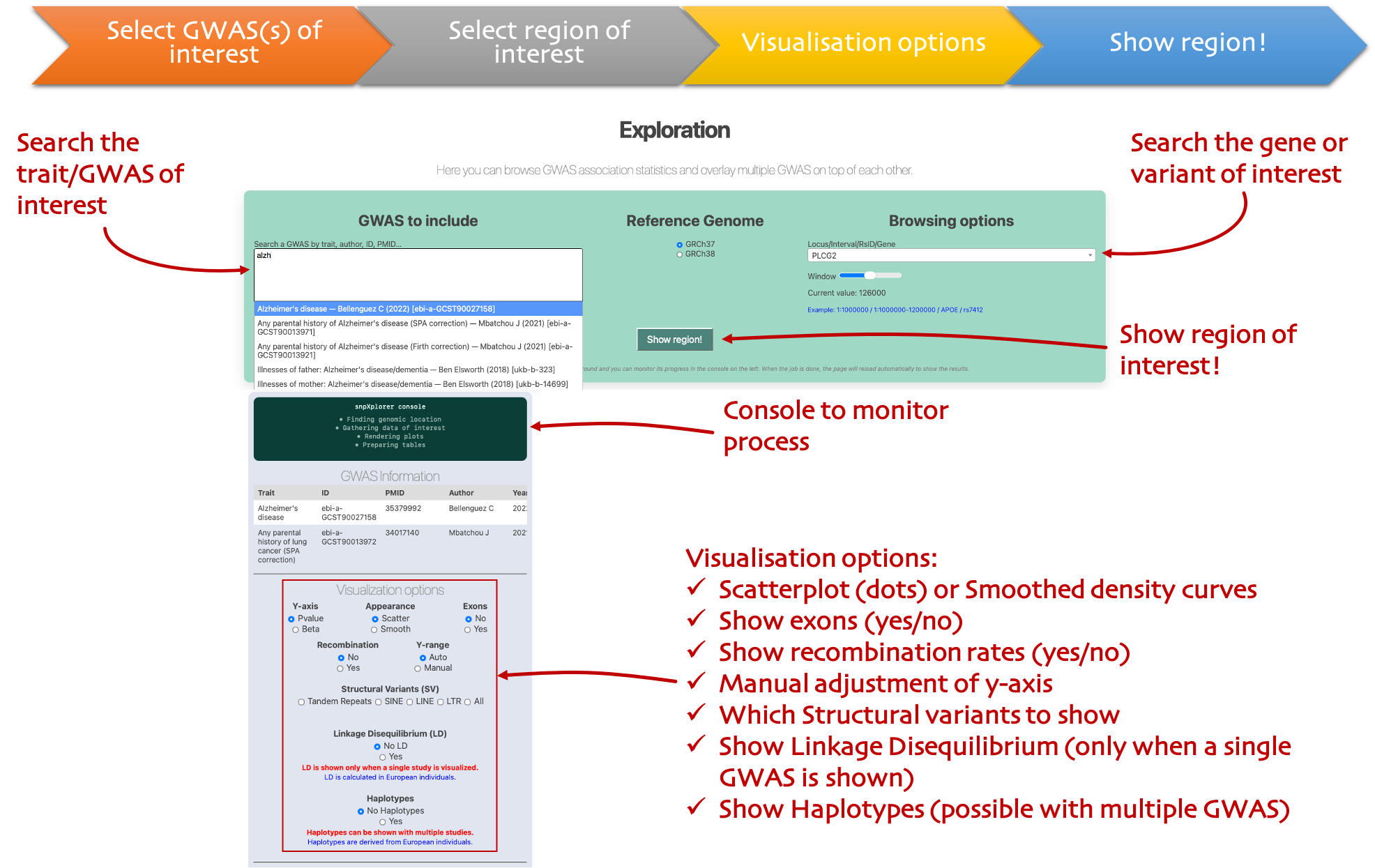
Exploration
Regional plots
Pick GWAS datasets, search a SNP/gene/region, and explore signals with LD, SVs and GTEx.
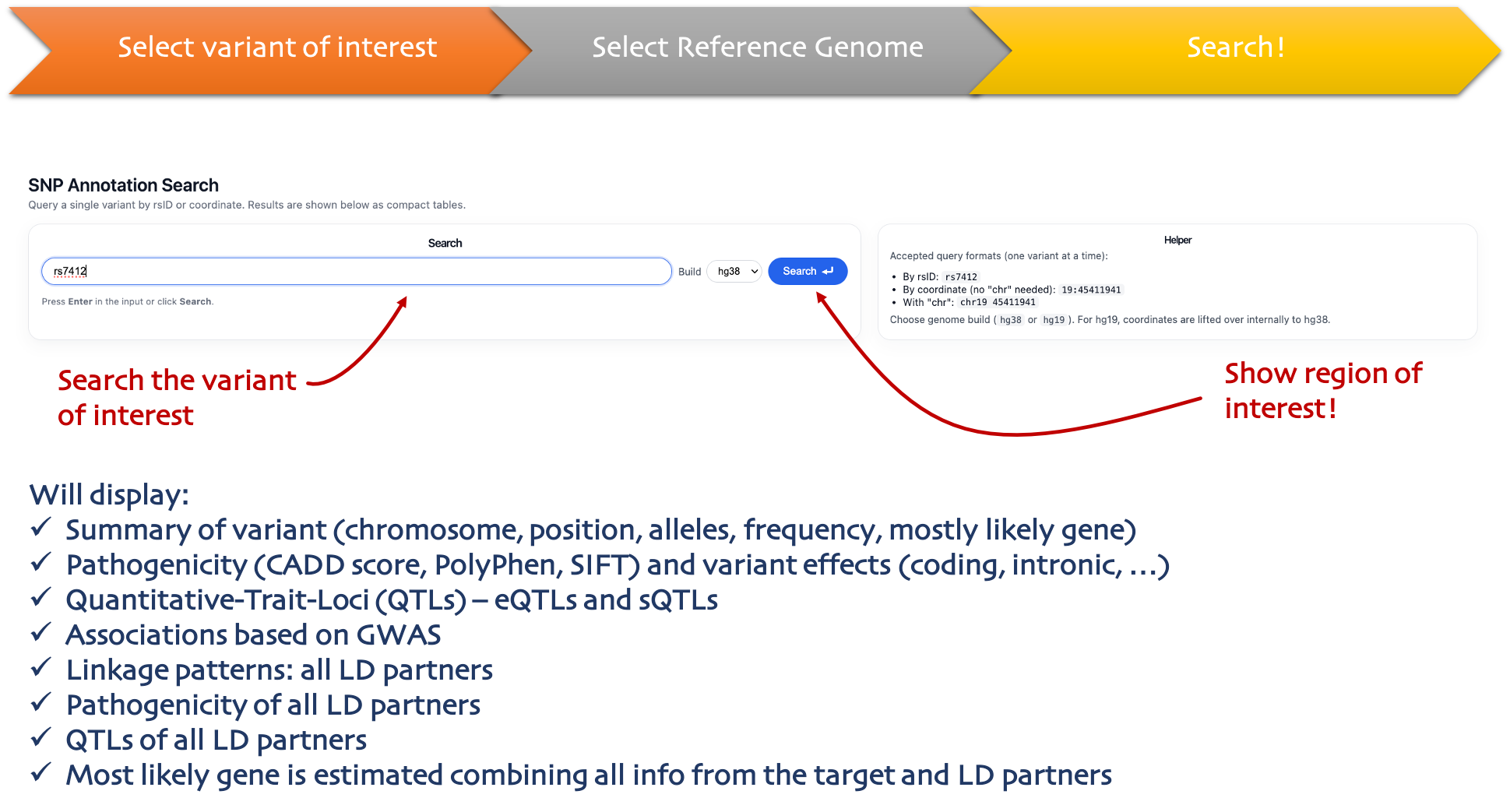
Single Annotation
One variant
Query one variant and see consequences, QTLs, GWAS links, and LD-proxy annotation.
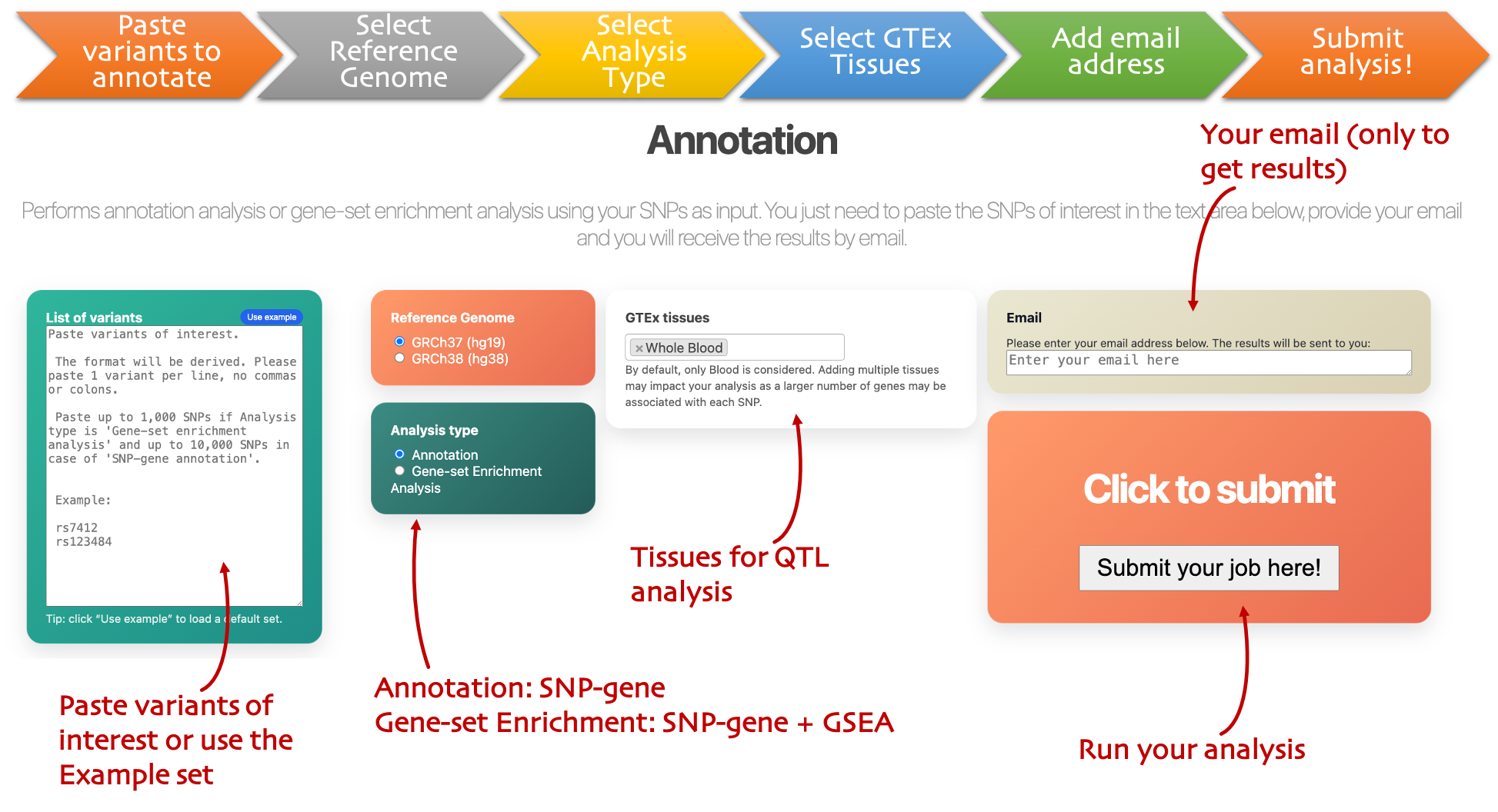
Batch Annotation
Many variants
Submit a SNP list for annotation, with optional gene-set enrichment and emailed results.
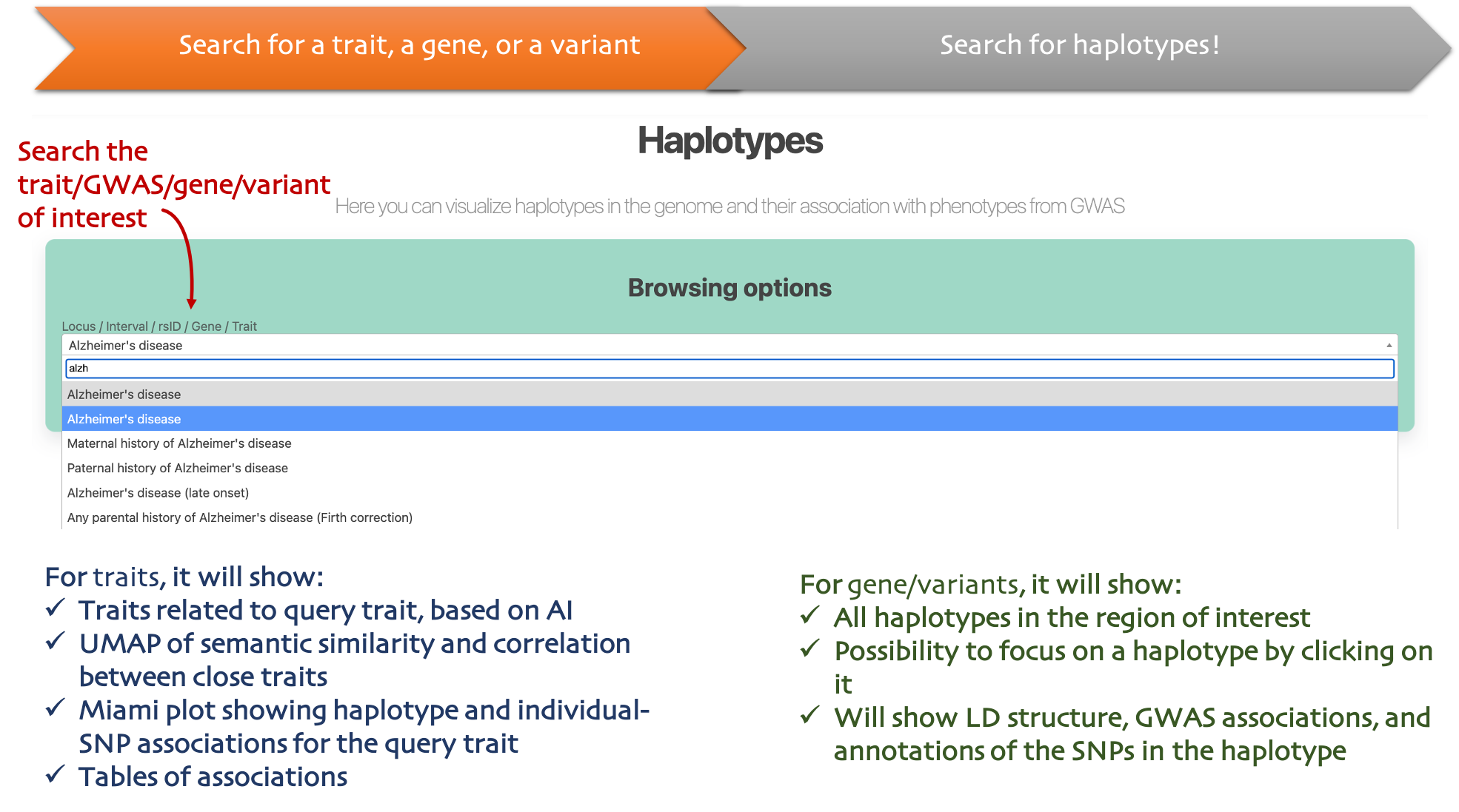
Haplotypes
Context
Explore haplotype structure around loci and compare associations across related traits.
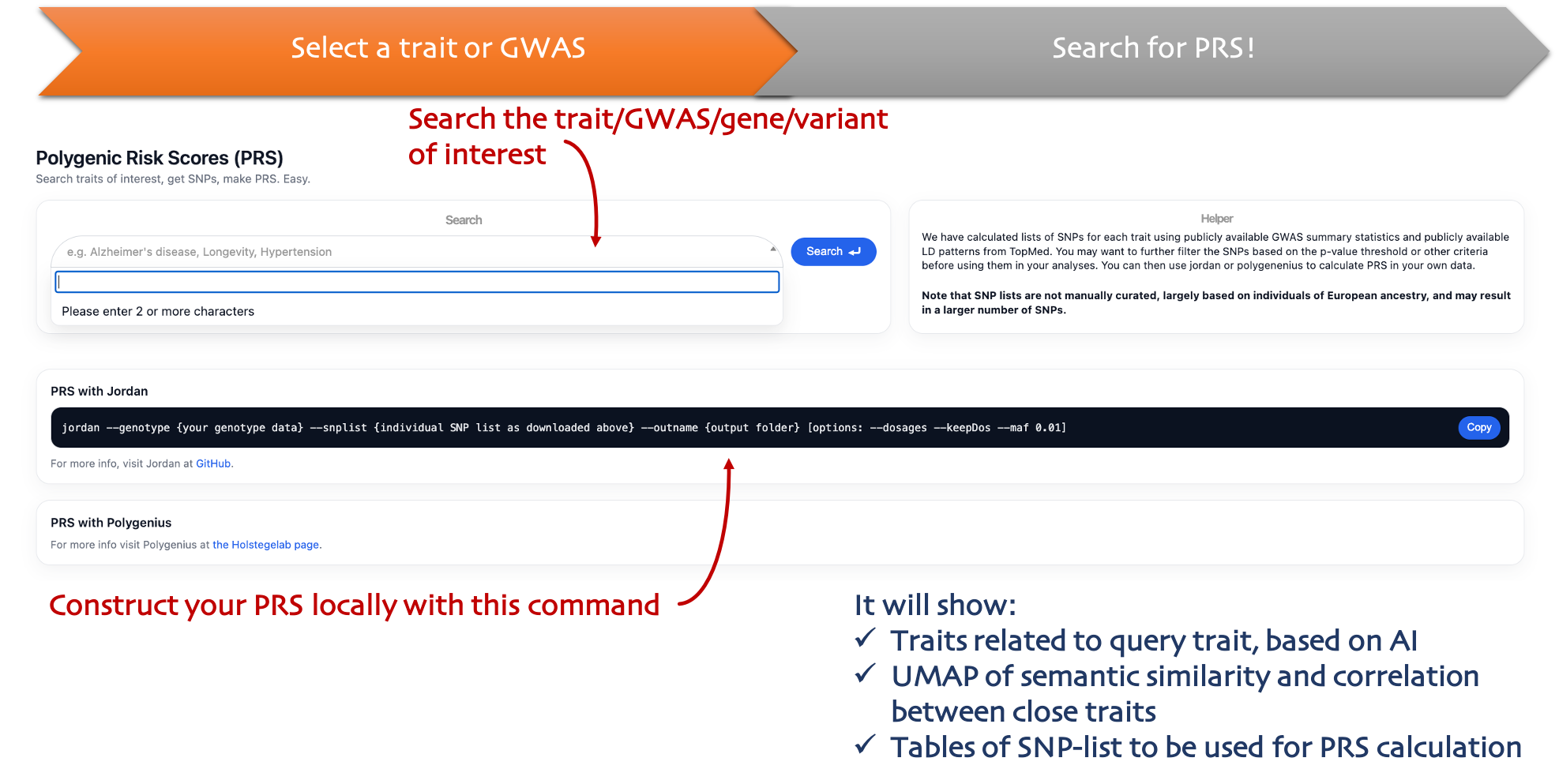
PRS
Variant sets
Browse curated variant sets for PRS construction (clumping+thresholding) and download SNP lists.


